Addressing persistent challenges in digital image analysis of cancer tissue: resources developed from a hackathon
Molecular Oncology, 2025.
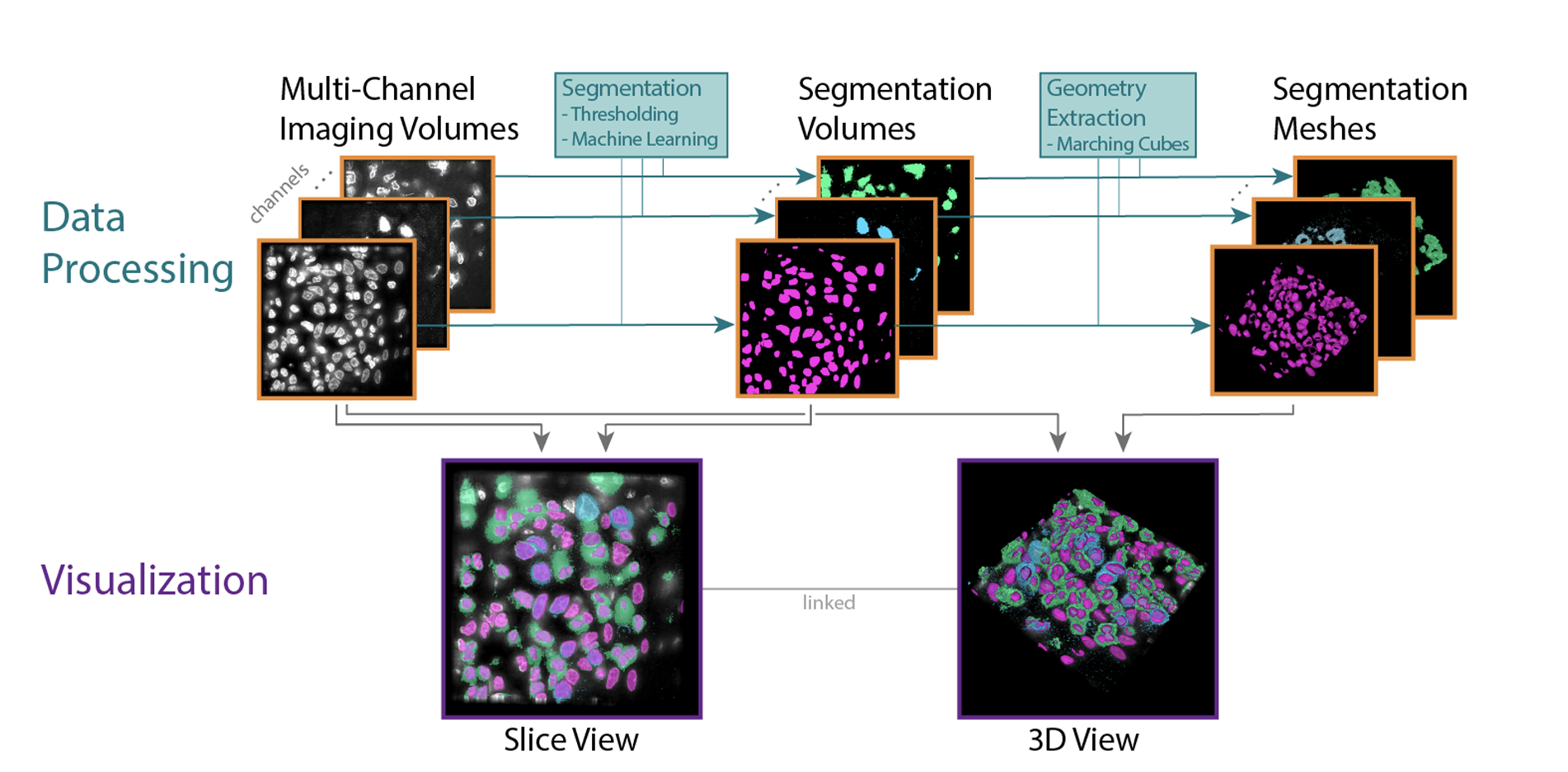
The National Cancer Institute (NCI) supports many research programs and consortia, many of which use imaging as a major modality for characterizing cancerous tissue. A trans-consortia Image Analysis Working Group (IAWG) was established in 2019 with a mission to disseminate imaging-related work and foster collaborations. In 2022, the IAWG held a virtual hackathon focused on addressing challenges of analyzing high dimensional datasets from fixed cancerous tissues. Standard image processing techniques have automated feature extraction, but the next generation of imaging data requires more advanced methods to fully utilize the available information. In this perspective, we discuss current limitations of the automated analysis of multiplexed tissue images, the first steps toward deeper understanding of these limitations, what possible solutions have been developed, any new or refined approaches that were developed during the Image Analysis Hackathon 2022, and where further effort is required. The outstanding problems addressed in the hackathon fell into three main themes: 1) challenges to cell type classification and assessment, 2) translation and visual representation of spatial aspects of high dimensional data, and 3) scaling digital image analyses to large (multi-TB) datasets. We describe the rationale for each specific challenge and the progress made toward addressing it during the hackathon. We also suggest areas that would benefit from more focus and offer insight into broader challenges that the community will need to address as new technologies are developed and integrated into the broad range of image-based modalities and analytical resources already in use within the cancer research community
Acknowledgements
The hackathon was generously supported by sponsors Carl Zeiss, Lunaphore Technologies, and MARK III Systems through prizes for hackathon winners. The authors, hackathon organizers, and participants are funded by NIH grants 3U54CA225088-04S1, U2C-CA233262, U54CA225088, U54CA209988, U2CCA233280, U54CA217450, R50CA243783, U54CA217450, NSF grant NCS-FO-2124179, NSF grant IIS-1901030. We would also like to acknowledge the excellent work performed by several of the participants who were also awarded prizes for their efforts during the hackathon, including Edward Novikov, Laurent Bataille, and Monica Del Valle. We would also like to acknowledge the contribution of several participants with whom we have lost contact and have been unable to include them as authors on the manuscript, including: Luke Sargent, Laurent Bataille, Walid Bousselham, Behnaz Bozorgui, Elmar Bucher, Chamika Gangul, Qiang Gu, and Jason Lu.